Task1 - Learning the Linux shell
This task will introduce students to the Linux command-line (shell environment).
Requirements
- Access to a linux-based OS running BASH
What is the Linux Shell?
The shell is a command-line programming language for interacting with the UNIX operating system.
There are several different shell languages. What we will be using in this course is a popular shell flavor called BASH
We will be learning basic commands, but BASH is actually a language that can perform complex programming tasks.
Getting Started
Before you can begin with the coding exercises, you must have access to a linux machine. You can either use your own local system or a remote VM that has been set up for you.
Accessing the remote VM
If you have been given access to a remote VM, you can access it either through the url that has been given to you, or directly through the Terminal like this:
ssh -i /path/to/your/.ssh/publickey yourUserName@remoteIP
When you are done, you can leave your session by typing…
exit
Once you are logged on | Learning the command-line
If you have logged in correctly, you should see a welcome screen.
You are now in a unix command-line environment. For more information and instructions:
- CodeAcademy - Learn the command line
- TeachingUnix - Another Unix Tutorial
- BasicCommands - List of common commands
A Linux Shell primer
Navigating files/folders
When you log in, by default you start in your home directory.
Type…
pwd
And this will print to the screen your current location (e.g., /home/username)
You can always get back to your home folder by typing
cd
To view the contents of your current folder type
ls
To make the folder “task1”, type
mkdir task1
Change directory into ‘task1’ folder
cd task1
And now create a file called file.txt
>file.txt
Open the file with nano. This is one built-in text editor. There are others.
nano file.txt
Now enter a few lines of text, type ‘ctrl-o’ and then ‘ctrl-x’ to save and exit
To print to the screen the contents of your file
cat file.txt
Other ways of viewing your file
less file.txt #type q to exit
more file.txt
head file.txt
head -n 10 file.txt # first 10 lines of your file
tail file.txt
tail -n 10 file.txt # last 10 lines of your file
Size of your file
du file.txt
du -h file.txt # in human-readable output (byte, kb, mb, etc.)
To count the number of words and lines in your file
wc file.txt #words
wc -l file.txt #lines
wc -m file.txt #characters
To copy the file to a new file
cp file.txt newfile.txt
You can also ‘move’ a file to a new location or rename it using mv.
To combine both files together into a third file
cat file.txt newfile.txt > thirdfile.txt
The ‘>’ redirects the output of the commands on the left of it to a file specified on the right.
Delete the file (note: be careful since there is no Trash Bin)
rm file.txt
Print contents of all .txt files in current folder. * acts as a wildcard
cat *.txt
Delete all .txt files in the current folder
rm *.txt
Delete all files in the current folder
rm *
Move back to the previous folder
cd ..
And delete the folder ‘task1’
rmdir task1
Getting help on linux commands and program usage
For most commands, you can get more information on their usage by typing man ‘command’.
e.g., try
man ls
#type 'q' to quit
#Note: this line and the line above are interpreted as comments since they start with the "#" character. They will not be executed as a command.
man will work with some bioinformatics tools. However, not always.
e.g., for help on the blastp tool, type
blastp -h # or
blastp --help
Additional operations
Pattern finding with grep
grep "word" file.txt # prints lines in file.txt containing "word"
grep -o "word" file.txt #print out all the occurrences of "word"
grep -c "word" file.txt # counts the number of lines containing "word" in file.txt
Note: Be careful when using grep to analyze files containing nucleic acid or protein sequence data.
e.g., your FASTA file may be separated into multiple lines like this:
>myFastaSequence
ATCGACGTTATCGACTAGCTAT
TCGGCGCGGTATTAGCGATTCG
TAATATCGGCGCGATATATCGA
instead of this:
>myFastaSequence
ATCGACGTTATCGACTAGCTATTCGGCGCGGTATTAGCGATTCGTAATATCGGCGCGATATATCGA
Therefore, grep may miss some words that span multiple lines.
Fortunately, there is a useful tool called compseq that be used to examine the k-mer composition of a FASTA file.
It can be run like this:
compseq file.fasta
#or
compseq -reverse file.fasta #also counts occurrences on the reverse complement of the sequence
Piping commands
We can also chain together multiple commands like this using the | (pipe) operator.
grep "word" file.txt | wc -l # will count the number of lines containing the word "word"
# or alternatively
cat file.txt | grep "word" | wc -l # does the same thing as above
# if we want to count ALL the occurrences of "word" in the file (allowing multiple per line), we can do
grep -o "word" file.txt | wc -l
Copying a file to and from a remote server
To
scp /path/to/file.txt username@remoteserver.com:/path/to/location/.
From
scp username@remoteserver.com:/path/to/file.txt /path/to/location/.
Uploading/downloading files via Google cloud’s ssh browser
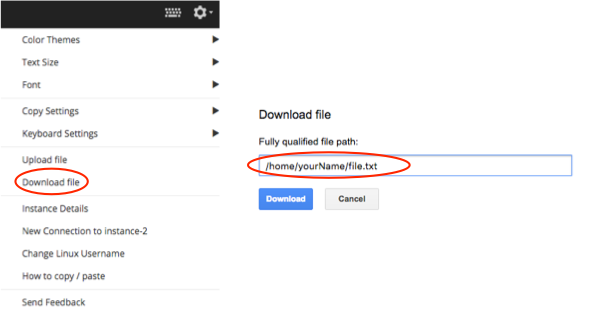
If you are using ssh in a browser window, you can easily upload and download files via the browser.
Look in the rop right corner for the options window, click Download file (see here) and then enter in the path to your file.
Remember, your current path can be found using pwd. A useful command for printing out the path to your file is:
{kind=link}
realpath file.txt
Downloading a file off the internet
wget <url>
File compression/uncompression
This is done using programs such as tar, and gzip and gunzip.
e.g.,
gzip file.txt # to compress it
gunzip file.txt.gz # to uncompress it
More tips
Use tab to autocomplete
Use tab for autocompletion! This will speed up your command-line work dramatically. More here: tab-autocomplete
Use Ctrl-C to interrupt or end a process
If you need to interrupt a command or process that you have started, press Ctrl-C.
Other tips for becoming a linux power user
ASSIGNMENT QUESTIONS
PLEASE COMPLETE ASSIGNMENT 1 ON LEARN. This can be found under Quizzes.
You will be asked to answer the following questions.
Hint: remember to use man if you want to explore added functionality of commands.
Download and uncompress this file containing the genome sequence of E. coli K12 https://github.com/doxeylab/learn-genomics-in-linux/raw/master/task1/e-coli-k12-genome.fasta.gz
 Q1 - What is the size of the uncompressed file in megabytes (round to one decimal place)?
Q1 - What is the size of the uncompressed file in megabytes (round to one decimal place)?
Q2 - How many characters are in this file?
Q3 - How many lines in the file contain the word “ATATATAT”?
Q4 - What character (A, C, G, or T) is most common in the file?
Q5 - What is the most common trinucleotide in the file? Hint: for an easy way to answer this, try the program compseq.
Q6 - How many times does the word “CCGGAG” occur in the genome sequence?
Q7 - What is the answer to the above question if you also include matches on the reverse complement of the genome sequence?